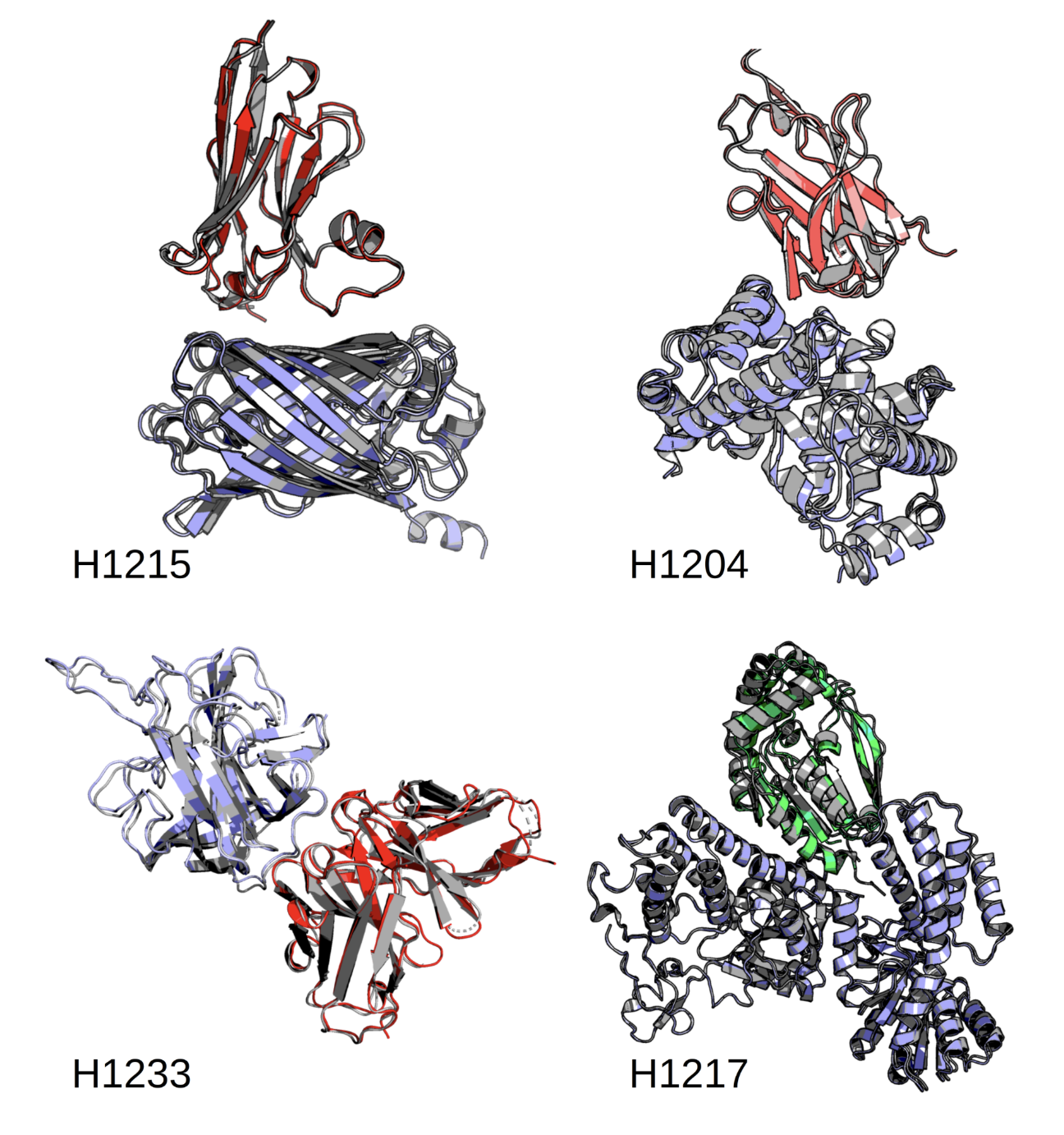
At the smallest scales of life, everything comes down to molecules. Proteins fold, shift, and interact in constant motion, carrying out the processes that sustain life. Drugs, too, are simply molecules, designed to bind, block, or alter these interactions. But understanding how these systems behave is far from simple. They are too small to see directly, too complex to capture fully through experiments, and too dynamic to describe as static structures.
The practical stakes are significant. Designing a drug depends on understanding how a molecule will bind to a specific protein target. Traditionally that process has required screening millions of compounds experimentally. Computational modeling offers a more targeted path, narrowing the field before anything is synthesized in a lab.
For Dzmitry Padhorny, who joined the Oden Institute for Computational Engineering and Sciences at The University of Texas at Austin as a research assistant professor in Fall 2025, this work builds on research he began at Stony Brook University in New York. His research addresses challenges at the intersection of mathematics, physics, and biology. He develops computational methods to model molecular structures and interactions, combining deep learning with physics-based approaches to better understand biological systems and how they can be manipulated.